Prodine-type Incapacitants
In the '60s, pharmaceutical companies searching for analgesics to replace morphine synthesized thousands of derivatives of 1-substituted-4-phenyl-4-piperidine. Four chemical groups of drugs have reached the clinical trial stage, typical representatives of these groups are Pethidine, Prodine, Ketobemidone, and Picenadol.
Pethidine (Meperidine) |
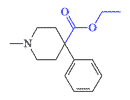
Prodine (Nisentil) |
Ketobemidone |
Picenadol |
In general, 4-phenyl-4-piperidine derivatives are not commonly used in medicine because they do not offer any advantages over morphine.
Pethidine-, Ketobemidone-, and Picenadol- type Analgesics
Pethidine (Meperidine). In the '30s, Germany produced the first wholly synthetic opioid, Pethidine. The chemical formula of Pethidine is structurally similar to morphine and atropine, so it is most effective for pain accompanied by smooth muscle spasms. Pethidine potency relative to morphine is 50% in most animals[19] but only 10% in humans — 10 mg of morphine is equivalent to 100 mg of pethidine.[18]
However, the activity of Pethidine derivatives can increase significantly, up to hundreds of times, if the phenyl ring is connected with the nitrogen atom by a straight chain of 3 carbon atoms.[5] The most potent drugs in this group, Phenoperidine and R 951, are 5–20 times stronger than morphine.[21], it was also, at the time of its synthesis, the most potent opioid in the world[41].
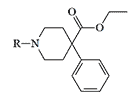 |
|||
| Non-proprietary Names | R | Activity in humans (Pethidine=1) |
Dosage and route of administration |
|---|---|---|---|
| Anileridine Leritine Alidine, Nipecotan, Adopol |
1–4 | 25–100 mg, intramuscularly[23] |
|
| Morpheridine TA 1 |
4 | 25 mg, intramuscularly[25] |
|
| Benzethidine | 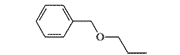 |
<5 | *AD27 — 20 mg, subcutaneously[24] |
| Pheneridine |
2–10 | 10–50 mg[33] | |
| Etoxeridine Atenos Carbetidine |
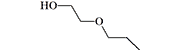 |
5–10 | 10–20 mg, intramuscularly[22] |
| Piminodine Alvodine, Cimadon, Win 14098-2 |
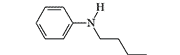 |
13 | 7.5 mg, subcutaneously[26] |
| Furethidine | 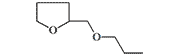 |
<20 | *AD33 — 5 mg, subcutaneously[24] |
| Phenoperidine R 1406 |
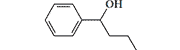 |
50 | 2 mg, intramuscularly[21] |
| R 951 | 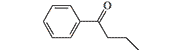 |
30–200 | 0.50–3 mg[33] |
| Analgesic activity of Pethidine derivatives in animal testing | |||
| R 1597 |  |
50 (mice) 228 (rats) |
subcutaneously[36] subcutaneously[36] |
| R 951 | |
275 (rats) |
subcutaneously[5] subcutaneously[5] |
| Phenoperidine R 1406 |
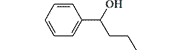 |
286 (rats) |
subcutaneously[5] subcutaneously[5] |
| 1-(7'-fluoro-n-heptyl-1')-3β-methyl-4-phenyl-4-carbethoxy-piperidine | 1400 (mice) | subcutaneously[27] | |
| 1- (6'-nitro-n-hexyl-1')-3β-methyl-4-phenyl-4-carbethoxy-piperidine | subcutaneously[27] | ||
*AD33 and *AD27 doses causing analgesia in 33% and 27% of patients, respectively
In 1965, Edgewood Arsenal studied the incapacitating effects of three meperidine-type compounds on monkeys, but the results were not disclosed.[42]
Ketobemidone, another German analgesic, was synthesized during World War II. It has been reported to possess analgesic activity comparable to morphine. Ketobemidone relieves severe pain when other opioids don't work but develops an intense physical dependence (addiction) with great rapid. It is banned in the USA but is still used in some Scandinavian countries.
The Central Intelligence Agency became interested in this group of analgesics even earlier than their colleagues in the U.S. Chemical Corps. The CIA needed effective knockout drugs for covert operations, and Prodine and related substances were deemed useful for this purpose. During the '50s, the CIA's behavior modification program MK-Ultra expressed interest in
”chemicals which have been found inactivate or permanently destroy specific portions of the nervous system. Ketobemidon is one such drug. It is used in (classified) to some extent in the treatment of drug addicts. Nisentil is related compound with effects which suggest a similar action. These compounds are ordinarily introduced by injection, although they would probably be effective if admin. as an aerosol.”[10]
Picenadol is a mixed agonist-antagonist opioid that often causes dizziness, confusion, speech disorders, and tremors in patients. Following a short clinical trial, its usage was discontinued.[31]
Prodine-type Incapacitants
The best-known drugs in this group are Prodine in the United States and Promedol in the former Soviet Union. These are older analgesics with inherent disadvantages of all opioids in the form of nausea, vomiting, itching, euphoria, and, most importantly, respiratory depression. Prodine was first synthesized by American chemists in 1947.[11] A year later, its Soviet analog, Promedol, was synthesized in the USSR. Today, Promedol is one of the most prescribed opioid analgesics in the former Soviet Union. Also, a unit-dose syringe with Promedol was part of the Soviet Army first aid kit until 2012.
The action of Prodyn is shorter than morphine, so it is more suitable for brief surgical procedures. Prodine was popular with pediatric dentists to sedate difficult child patients before a procedure, but after several deaths, the manufacturing firm withdrew it.[32]
Analgesic activity of Prodine and Promedol is relatively low, ranging from 15–50% of morphine activity. However, later, substances hundreds and even thousands of times more powerful were discovered. In large doses, prodine derivatives induced unconsciousness in humans and could be used as non-lethal chemical agents (incapacitants).
In 1943, Janssen et al. found that analgesic activity increases up to 5-fold when carbethoxy in Pethidine-type opioids (–COOC2H5) is replaced by propionoxy (–OCOC2H5) in prodine-type drugs.[30] In the laboratories of P. Janssen, B. Elpern, and N. Eddy synthesized derivatives of 4-phenyl
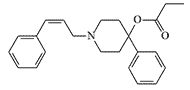 |
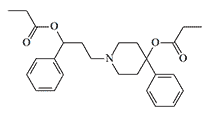 |
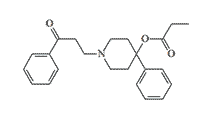 |
| Compound Elpern X785 pethidine[9] |
Compound Janssen X1500 pethidine[1] |
Compound Carabateas (OPPPP) X1350 pethidine[1] |
In 1958, CIA contractor H. Isbell met with famous Belgian pharmacologist P. Janssen. In conversation, P. Janssen informed his colleague that he had succeeded in synthesizing new drugs with remarkable analgesic activity:
"One of Janssen's compounds is said to be 3-thousand times as potent as morphine in monkeys; and another, a rather unstable fluorinated compound, is 300-thousand times as potent as morphine. I believe that Dr. Janssen talked with Dr. N. B. Eddy and with persons from the Army Chemical Center concerning these drugs. It is quite likely that members of this series will come to us via the Drug Addiction Committee after testing in monkeys in Michigan has been completed."[12]
H. Isbell headed Project MKPILOT, which aimed to find potent opioids that the CIA could use to immobilize humans. From 1967–73, the CIA partnered with Edgewood Arsenal on the secret MKCHICKWIT program, its objective was "to identify new drug developments in Europe and Asia and to obtain information and samples."[13] But obviously, the US Army and CIA's collaboration with European pharmacologists began much earlier.
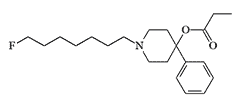
P. Janssen never mentioned it in his publications about this super-powerful analgesic, but perhaps it was the candidate chemical agent EA 2230, which caused immobilization in mice at extremely low doses but proved too toxic for primates.[4] These Prodine-type incapacitants, generally referred to as prodines, were developed at the U.S. Army Chemical Research, Development and Engineering Center (CRDEC) until at least 1975, and possibly later.[4]
Table 2. Prodine-analogue incapacitants tested in the '60s
| EA 2219 | |
R1317, WIN 14465/2, NIH 7718. First synthesized in 1958, in mice it is 100 times more analgesic than morphine.[9] Administration to monkeys of 0.01-0.02 mg/kg causes a drop in blood pressure, and 0.03 mg/kg leads to respiratory arrest.[4] |
| EA 2221 | 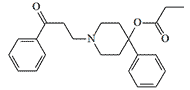 |
OPPPP. One of the most active prodyn derivatives, EA 2221, at a dose of 0.005 mg/kg, induces prostration in monkeys. However, the lethal dose is only 6 times the effective dose.[4] |
| EA 2228 | 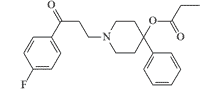 |
Minimum effective dose in monkeys is 0.01 mg/kg, lethal dose is 0.03 mg/kg. |
| EA 2230 | 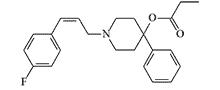 |
EA 2230 induced immobilization in mice at a minimal effective dose (MED50) 0.00032 mg/kg, but to induce prostration in monkeys required a 100-fold higher dose.[4] |
| EA 2392 | 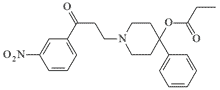 |
At a dose of 0.01 mg/kg it induces prostration in monkeys. |
| EA 2475 | 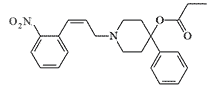 |
In experiments on mice, EA 2475 was far superior to carfentanil (MED50 — 0.000056 mg/kg), which was still unknown at that time, and had a fantastic therapeutic index — more than 50 000, higher than that of remifentanil, considered the safest opioid. In reality, it has proven to be ineffective and toxic to primates (MED50 — 0.1 mg/kg).[4] |
| EA 3382 | Formula unknown | Dart gun paralysant for clandestine ops. Used to equip special weapons that shoot poisoned darts. |
| EA 3407 | 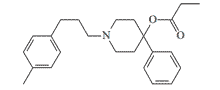 |
EA 3407 causes immobilization of mice at a minimum effective dose (MED50) of 0.006 mg/kg and has a high safety index (LD50/MED50) of over 5000, but is ineffective and extremely toxic to monkeys. |
Like other opioids, prodine derivatives have shown incredible activity and low toxicity in rodent experiments. However, in primate experiments, fatal respiratory arrest occurred at doses only a few times the analgesic dose.
The search for a safer incapacitant led to substances with heterocyclic substituents instead of phenyl. The 2-thienyl analog EA 2221 had an 8-fold higher margin of safety (LD50/MED50) than EA 2221. Interestingly, replacing the sulfur atom with oxygen resulted in an almost 30-fold increase in toxicity, and this substance is considered a potentially lethal chemical agent.[2,3]
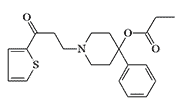 Incapacitating Chemical Agent |
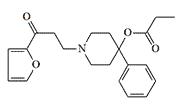 Lethal Chemical Agent |
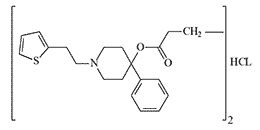 |
The safest known 2-thenoylethyl derivative, like many other opioids, causes an increase in chest muscle tone, referred to colloquially as “wooden” chest. In CRDEC, scientists developed prodine-like incapacitants with a chemical structure similar to peripheral myorelaxants. These compounds, when tested, gave reaction signs of relaxation of the skeletal muscles in experimental animals, but their biological activity was too low.[7]
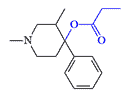
The addition of a 3-methyl group to the piperidine ring significantly enhances the activity of prodine derivatives. The strongest prodine-type analgesic, according to P. Janssen, is a 3-methyl analog of OPP/PPP, which is 1000 times more potent than morphine. There are eight isomers in this substance, and there may be even more active analgesics among them.[40]
 Compound Janssen X1000 morphine |
 Compound Fancher X150 morphine[28] |
 Compound Boehringer-Ingelheim X450 morphine[29] |
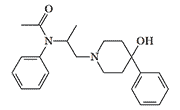
CRDEC also studied derivatives of 4-phenylpiperidinol not containing propionoxy group, among which there are analgesics more potent than morphine. One such compound, synthesized by O. Fancher et al. in 1964, was 150 times more potent than morphine.[28] This analgesic contains a fragment of the fentanyl structure, causing unusually high activity. However the removal of the propionic radical strongly reduced the activity of the experimental chemical agents listed in Table 2.[4]
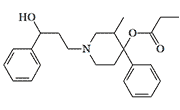
It is believed that adding a long carbon chain to the nitrogen of 4-hydroxy-substituted piperidines would cause their analgesic activity to disappear. The compounds synthesized by pharmaceutical company Boehringer-Ingelheim, however, break this rule — their activity is a several hundreds times higher than that of morphine.[29]
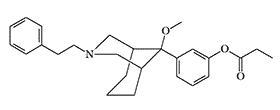
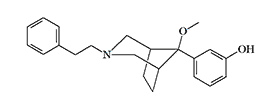
Between 1982 and 1989, a team of Chinese pharmacologists from the Shanghai Institute of Materia Medica conducted research on the pharmacology of azabicyclononanes and azabicyclooctanes. These are very powerful bicyclic analogs of prodine (see azabicyclononanes).
 |
 |
|
P-7548 |
Azabicyclooctane |
Opioid/Deliriant Combination. The combination of opioids and anticholinergic drugs has been used since the 9th and 10th centuries to induce anesthesia in surgical patients. Opium and mandrake juice, which contained the alkaloid scopolamine, were part of Spongia somnifera ('soporific sponge'). Drug substances were soaked into a sponge and placed on the patient's nostrils to produce deep sleep.[15] The combination of morphine and scopolamine was popular among obstetricians in the early 20th century. In addition to providing pain relief, it induces sedation and indifference while keeping laboring women conscious with limited memory of the moment of labor.[16]
In the 70s, Edgewood Arsenal studied a two-component incapacitant consisting of an opioid analgesic EA 3382 and a chemical agent from the group of anticholinergic drugs EA 3443, similar in action to scopolamine. When drugs of these two groups are used together, the depressing effect on CNS should be mutually enhanced, and undesirable side effects should be reduced by decreasing the dose of each component.
In animal experiments, opioids co-administered with a cholinergic-blocking drug caused prolonged but reversible paralysis as a result of deep CNS depression. It was expected that these substances would have a similar effect on humans, loss of consciousness was supposed to occur in 10–20 minutes and last for several hours. However, trials on humans were never completed due to the risk of seizures.[8]
Opioid/Opioid Antagonist Combination. Administering prodine and opioid antagonists simultaneously prevented respiratory depression and preserved pain relief.[17] The combination of prodine EA 3382 with a benzomorphan derivative CAR 220,548 (which acts as a partial antidote and is also a potent hallucinogen) has been studied in the USA as a potential incapacitant.[8]
By the late '60s, hundreds of prodine derivatives had been synthesized, many of which were several orders of magnitude superior to morphine, but despite some encouraging results obtained in animal experiments, CRDEC was subsequently favored over fentanyl and its derivatives, which had a more attractive pharmacological profile.
Designer Drugs
Strong opioids such as carfentanil, which can be purchased on the Internet in quantities of several kilograms, can represent a serious terrorist threat. This also applies to derivatives of Prodine, which are easier and cheaper to synthesize than fentanyl. For example, the acetate homolog of MPPP is produced in two steps from readily available precursors such as methylamine, formaldehyde, acetic acid, and α-methylstyrene.[38]
MPPP (MethylPhenylPiperidinylPropionate) — is the first of the prodine-type "new heroins." Two to three times stronger analgesic than morphine.[6] Its chemical structure is Desmethylprodine, street name "Super Demerol." Synthesized by an amateur chemist trying to create a legal alternative to heroin in 1979. The chemist soon developed symptoms of Parkinson's disease. It was discovered that some of the samples he had produced contained as much as 97% neurotoxin MPTP.[34]
PEPAP (PhenEthylPhenylAcetoxyPiperidine). The analgesic and euphoric activity of MPPP and PEPAP is equivalent to 0.7 of that of heroin.[37]
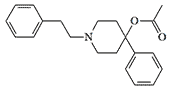
PEPAP |
OPPPP |
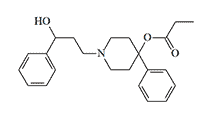
OPP/PPP |
OPPPP (OxoPhenylPropylPhenylPiperidinylPropionate) is the same substance listed in table 2 under code name EA 2221. PEPAP and OPPP was seized several times in the USA and Canada in the '80s. The analgesic and euphoric dose of OPPPP for humans is estimated at 0.1 mg.[6]
OPP/PPP (OxyPhenylPropyl/PhenylPropionoxyPiperidine). In 1988, a raid on a clandestine laboratory resulted in Drug Enforcement Administration (DEA) agents discovering a batch of drugs called in the media as PEPAP.[35] But as it later turned out, the seized substance was a previously unknown designer drug called OPP/PPP.[37] OPP/PPP was first synthesized by Carabateas in 1962, it was 3200 times more potent than Pethidine in animal experiments.[1]
This is how a DEA agent describes the effects of this synthetic heroin substitute:“... causes an immediate effect that includes a heroin-like euphoria “but with a more dreamlike or spacey sensation typical of PCP. There’s a disorientation, visual illusion, auditory hallucinations and the blurring of vision.”
“This stuff is super-deadly, it is so potent in its pure form that even fumes can kill... It is hard to dilute down to useable strength.”[35]
According to the DEA, a synthetic heroin substitute that is 36 times more powerful than pure heroin.[35] The entire manufactured batch of drugs was confiscated and did not end up in the hands of drug dealers.
Parkinsonian toxin MPTP
In the 70s and 80s, synthetic prodine-type heroin surrogates were being identified in small batches of designer drugs. But in general, despite the simple and affordable synthesis, drugs of this group do not attract the interest of clandestine chemists, probably because of the danger of contamination of the final product with the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), which causes Parkinson's-like symptoms in humans.[6]
In 1982, in California, an unusual disease associated with injections of "bad heroin" began to occur among drug addicts. The diseased person fell into a strange frozen state — all his movements gradually slowed down, and after a while, he became completely immobile, his body stopped obeying him, but he remained conscious. He could see and hear everything that was going on around him, but he could not move or utter a word.[34]
There was even an urban legend among addicts that embalming fluid stolen from a nearby morgue was added to heroin. They called the strange condition "the walking death." However, the real culprit behind the tragedy was the MPTP impurity in the "bad heroin."[34]
In fact, the "bad heroin" was a mixture of heroin and the cheaper synthetic opioid MPPP produced in a clandestine laboratory. Due to improper reaction conditions, MPPP was replaced by the predominantly toxic MPTP. The formation of MPTP occurs in two ways: elimination of propionic acid from MPPP or dehydration of the intermediate 1-methyl-4-phenyl-4-piperidinol.
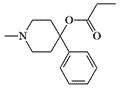
MPPP |
 |
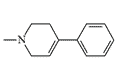
MPTP |
 |

MPP+ |
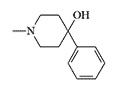
1-methyl-4-phenyl-4-piperidinol |
 |
In humans, MPTP is oxidized to MPP+ (1-methyl-4-phenylpyridinium) by monoamine oxidase B (MAO-B). MPP+ destroys cells in a small area of the brain known as the substantia nigra. The same death of the cells in the substantia nigra is also characteristic of Parkinson's disease.[39]
MPTP is an extremely toxic substance — the lethal dose for monkeys is about 1 mg/kg. Lower doses of MPTP cause catatonia in primates and humans: movements slow to a complete standstill and the person may freeze in an awkward posture for long periods, eyes stare ahead without blinking, speech is slurred, memory and reasoning abilities are also impaired. MPTP can be toxic if inhaled or in contact with the skin.[34]