Neuroleptics
Neuroleptics are a class of pharmacological agents used in psychiatry for the treatment of various mental disorders. In healthy individuals, they produce central nervous system depression and motor disturbances. Their principal advantages over other incapacitating agents are high biological activity combined with relatively low toxicity,[10] as well as a pronounced capacity to potentiate the effects of other non-lethal chemical agents, including fentanyls, adrenomimetics, benzomorphan psychotomimetics, and benzodiazepines. During the 1960s and 1970s, under the U.S. non-lethal chemical weapons program, experiments were conducted with two chemical classes of neuroleptics—phenothiazines and butyrophenones. In the Soviet Union during the same period, three areas of incapacitant development were considered priorities: neuroleptics, LSD, and BZ-type agents.[21]
The antipsychotic effects and motor disturbances produced by neuroleptics are attributable to blockade of D2-dopamine receptors, while the sedative effect is thought to be mediated largely through α2-adrenomimetic activity. In phenothiazine-class neuroleptics, sedation is more pronounced due to additional binding at M1-muscarinic and H1-histamine receptors.
Clinical Picture of Acute Neuroleptic Intoxication
Despite decades of clinical experience with neuroleptics in psychiatry, relatively little is known about their effects in healthy individuals—knowledge derived primarily from a limited number of volunteer experiments and from accounts of prisoners and psychiatric patients who were victims of punitive psychiatry in the United States and the Soviet Union.
Neuroleptic incapacitating agents can render a person combat-ineffective through two distinct mechanisms: disruption of higher mental function and induction of extrapyramidal symptoms.
Psychiatric effects. Neuroleptics exert a general suppressant effect on mental activity, blunting both cognitive and motor performance—effects that, when these agents are employed as incapacitants, should produce complete or partial loss of combat effectiveness. They produce powerful sedation which, at higher doses, transitions into somnolence. In patients with psychomotor agitation, a sense of calm emerges, internal tension subsides, drives are suppressed, and a state of psychomotor retardation sets in, characterized by profound indifference to the surrounding environment. In healthy individuals, however, neuroleptics paradoxically and frequently produce the opposite reaction—ranging from mild dysphoria to agitation and frank aggression.[28] Marked disturbances in logical thinking and difficulty performing simple tasks or sequences of actions may also be observed.
| Your thoughts are broken, incoherent; you can't hold a train of thought for even a minute. You're talking about one subject and suddenly you're talking about another. You start to roll a cigarette, drop it, pick up a book, take a shit, forget to wipe your ass. Your mind is like a slot machine, every wheel spinning a different thought. |
| (Description of the effects of fluphenazine by a California prison inmate)[36] |
| Your gaze cannot settle on anything; thought cannot take any definite form; even a visit from relatives becomes a burden. …The world turns empty and pointless. An inexplicable, unmotivated fear moves into the soul. The state is horrifying and almost beyond description. |
| (From the memoirs of a former Soviet political prisoner, on his reaction to a neuroleptic)[30] |
Extrapyramidal disorders. Neuroleptics disrupt the function of the extrapyramidal system—the brain structures responsible for regulating movement, muscle tone, and posture. The clinical manifestations are highly varied, but in acute intoxication the most commonly encountered are dystonia and akathisia.
Dystonia consists of spastic or repetitive contractions of muscles of the head, trunk, or limbs. Neuroleptic-induced dystonia characteristically presents with the following features:
- Backward or lateral deviation of the head (retrocollis and torticollis, respectively)
- Spasm of the masticatory muscles (trismus, forced yawning, grimacing)
- Slurred and effortful speech, tongue protrusion
- Spasm of the extraocular muscles (oculogyric crisis)[31]



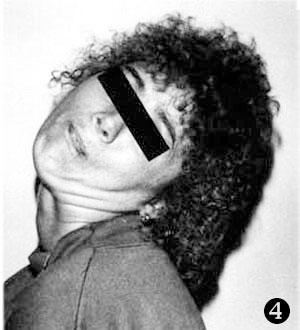

Various manifestations of dystonic disorders: 1) torsion dystonia; 2) reaction to fluphenazine administration presenting as retrocollis and oculogyric crisis; 3) acute dystonic reaction to haloperidol; 4) spasmodic torticollis; 5) involuntary tongue protrusion.
In the 1970s, neuroleptics were used in American prisons as a behavioral control measure, frequently without the inmates' consent.[36] In the Soviet Union, they served as one of the instruments of punitive psychiatry against political dissidents; accounts of haloperidol-induced dystonia can be found throughout the memoir literature of that period:
| There goes a prisoner down the aisle between the bunks, freshly given a handful of small white haloperidol tablets: akathisia is driving him on, his eyes glassy. And then you notice his jaw dropping involuntarily; he pushes it back up while he still can, but it falls further and further, and his muscles can no longer hold the strain. His mouth gapes wider and wider; he tries to force the jaw back with his hands, but in vain—in his bulging eyes there is pure terror. The spasm does not pass, and the jaw muscles stretch to their full extent, causing unspeakable pain. Then another spasm begins: the eyeballs roll upward, disappearing under the brow ridges, and from there the convulsion spreads through the entire body. The head is wrenched back, the fingers curl, the feet turn inward. The poor wretch collapses to the floor, groaning and moaning, thrashing like a wounded animal. His spine arches forward—this is called opisthotonus.[30] |
Akathisia (from Greek a—"not" and kathizein—"to sit"), or motor restlessness, is characterized by episodes of psychomotor agitation in which the affected individual experiences an irresistible compulsion to move continuously or to change body position without pause. The principal symptoms of akathisia include:
- Fidgeting and rocking
- Shifting weight from foot to foot
- Continuous walking in an attempt to relieve the inner restlessness
- Inability to remain seated or standing still for more than a few minutes[31]
The following account of akathisia was given by former Soviet political prisoner A. Shatravka:
| At first a person experiences an inexplicable restlessness. You sit down—and suddenly you want to stand up. You start walking—and suddenly you want to stop, or lie down. You lie down—and you can't stay there long either. You tear yourself off the bunk and start pacing the ward for no reason you can name. And so it goes until you are completely exhausted…[30] |
The neurological symptoms are frequently accompanied by psychiatric manifestations, including agitation, a sense of internal tension and anxiety, and insomnia.[4]
Neuroleptic parkinsonism, presenting as tremor, akinesia, and muscular rigidity, is uncommon in acute intoxication.
Phenothiazine-Class Incapacitating Agents
Phenothiazines constitute the largest and most thoroughly studied class of psychotropic drugs. Chlorpromazine, the first neuroleptic, revolutionized the treatment of schizophrenia in the 1950s—marking the first time that pharmacological agents were able to replace such barbaric psychiatric interventions as electroconvulsive and insulin coma therapies. Their severe adverse-effect profiles, however, caused patients no less suffering than the conditions they were meant to treat.
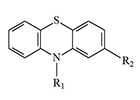 |
R2 | R1 | Name, synonyms |
Military code |
|---|---|---|---|---|
| –Cl | 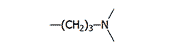 |
Chlorpromazine Thorazine Aminazin |
CS 4949 | |
| –Cl |  |
Prochlorperazine Compazine |
— | |
| –SCH3 | 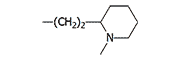 |
Thioridazine Sonapax |
— | |
| –Cl | 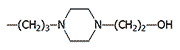 |
Perphenazine Trilafon |
— | |
| –CF3 | Fluphenazine Prolixin |
EA 5202 | ||
| –NO2 | — | EA 5432 | ||
| –CN | — | EA 5444 |
Perphenazine is 5–10 times more potent as a sedative than chlorpromazine[39] but is less toxic. Its acetate ester (etaperazine acetate) is ten times more active than the parent compound.[13] It possesses a pronounced antiemetic effect, including in cases of radiation injury. In the Soviet Union it was marketed as Aethaperazinum and was included in the AI-2 individual medical kit, and later in its successor, the AI-4.
Prochlorperazine. In 1970–1971 it underwent volunteer trials at the Edgewood Arsenal Biomedical Laboratory alongside fluphenazine and chlorpromazine.[20] As a neuroleptic it is 10–20 times more potent than chlorpromazine.
Thioridazine (Sonapax). In Russia during the 1990s, it was used by criminal organizations to immobilize victims during kidnapping, transport, and confinement. High doses produce a passive, stuporous state resembling deep sleep, accompanied by hypotension, tachycardia, and mydriasis. It was frequently administered mixed with alcohol, which reduced the likelihood of paradoxical psychomotor agitation.[3] Compared to other phenothiazine neuroleptics, it causes adverse effects considerably less often.
Chlorpromazine (Thorazine; CS 4949; 400,225). Marketed in the Soviet Union as Aminazin. Its capacity to suppress aggressive and defensive behavior in both humans and animals, combined with a pronounced sedative effect, repeatedly attracted the attention of military, law enforcement, and intelligence agencies. In 1954, the CIA funded Subproject 38 of the MKULTRA program, a classified investigation into the effects of chlorpromazine on human subjects.[4] Under a separate CIA contract, the Canadian psychiatrist Donald E. Cameron conducted research from 1957 to 1960 into behavioral modification techniques including electroconvulsive therapy, sensory deprivation, LSD administration, and prolonged pharmacological sleep. To induce a comatose state, Cameron used a drug cocktail that included chlorpromazine.
In 1993, David Koresh, the leader of the Branch Davidian sect, barricaded himself and his most devoted followers inside their compound against law enforcement. The standoff lasted nearly two months. Among the options considered, the FBI reportedly contemplated the use of a chlorpromazine solution in dimethyl sulfoxide (DMSO).[18] Phenothiazines penetrate the skin readily even in alcoholic solution, and the addition of DMSO was intended to further accelerate and enhance transdermal absorption. Ultimately, the decision was made to deploy tear gas grenades. During the final assault, an intense fire broke out—whether set by the sect members themselves or ignited by the grenades remains disputed. Seventy-six people died in the fire, including 21 children.
By the mid-1960s it had become clear that chlorpromazine's effective dose was too high and its toxicity too great for deployment as a non-lethal chemical weapon. Subsequent research was accordingly redirected toward identifying more potent and safer phenothiazine incapacitants, with fluphenazine and its derivatives as the primary candidates.
Fluphenazine (EA 5202) is approximately 20 times more potent than chlorpromazine in clinical terms and produces a less pronounced adverse-effect profile.
Despite more than a thousand papers on fluphenazine published in the 1960s, none described its effects in healthy subjects. Between 1965 and 1970, the Edgewood Arsenal Medical Research Laboratory conducted large-scale trials involving fifty healthy volunteers. The principal CNS findings were dysphoria, reduced activity, and diminished motivation.[11] Extrapyramidal symptoms occurred in more than half of all cases, appeared no earlier than 24 hours after administration, and included tonic spasms of the muscles of the neck, tongue, back, legs, and jaw. Fluphenazine proved more potent than its competitor haloperidol: its minimum effective dose (MED50) was 0.005 mg/kg, and the estimated median incapacitating dose (ID50)—the dose calculated to produce extrapyramidal disorders in 100% of cases—was 0.1 mg/kg.[11]
The psychiatric effects of fluphenazine in healthy volunteers proved entirely unexpected to the Edgewood researchers. In addition to the anticipated sedation, subjects exhibited heightened anxiety, irritability, and frank aggression—findings that substantially diminished fluphenazine's value as a potential incapacitant. At doses of 0.035–0.04 mg/kg, the most frequently observed symptoms were somnolence (89%), irritability (44%), agitation (33%), and blurred vision (33%).[28]
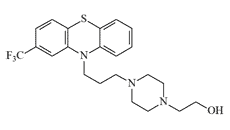
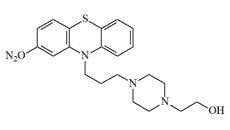
In 1971, researchers at the Edgewood Research, Development and Engineering Center (ERDEC) synthesized and evaluated fluphenazine derivatives in which the trifluoromethyl group at the 2-position was replaced by –NO2 or –CN. In animal studies these compounds proved effective and rapid-acting incapacitants; the 2-nitro derivative was even marginally more potent than the parent fluphenazine.[6]
 |
 |
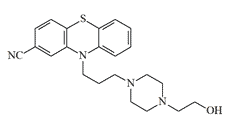 |
| EA 5202 (Fluphenazine) | EA 5432 | EA 5444 |
Field trials involving thermal aerosolization of the new incapacitants were conducted in 1974,[8] and in 1975 ERDEC developed an improved synthetic procedure for EA 5202 and its derivatives.[1,6]
Phenothiazine derivatives are known to irritate the respiratory tract and skin. To amplify this effect, ERDEC investigated replacing fluphenazine's trifluoromethyl group with a dicyanomethylene substituent –CH=C(CN)2, analogous to that in CS, and with a nitrovinyl group –CH=CH–NO2, analogous to that in the nitrostyrene-class irritants. This structural modification was intended to confer the irritant properties characteristic of "police gases" while retaining neuroleptic activity; however, the resulting compounds were significantly less potent as antipsychotics than fluphenazine.[6]
Butyrophenone-Class Incapacitating Agents
An outstanding contribution to the development of non-lethal chemical weapons was made by the eminent Belgian scientist Paul A. Janssen, founder of the internationally renowned Janssen Pharmaceutica. In addition to discovering the most potent incapacitants of the prodine, fentanyl, and carfentanil classes, Janssen and his colleagues were the first to synthesize most of the butyrophenone neuroleptics known today, several of which were evaluated as potential incapacitants: azaperone (1958), haloperidol (1958), spiroperidol (1961),[25] and 3-methylspiroperidol (1962).[26]
Azaperone is a veterinary neuroleptic used in animal husbandry to suppress aggression[22] and, in combination with narcotic or sedative agents, for temporary immobilization. The dose required for temporary immobilization in animals is relatively high—0.5–2 mg/kg—but when combined with fentanyl or etorphine it can be reduced to 0.05 mg/kg or less.
Haloperidol (CAR 302,034). The first volunteer trials of haloperidol were conducted by ERDEC in 1965 and yielded unimpressive results. At doses up to 0.03 mg/kg, haloperidol failed to produce complete incapacitation; peak effect was delayed by 8–12 hours; and the duration of action was excessive, persisting for up to 36 hours. Spastic muscle contractions appeared 12–16 hours after administration but were rapidly reversed by diphenhydramine.[11]
The effects of higher doses of haloperidol (0.125 mg/kg) in healthy volunteers were studied in 1985. At this dose, 75% of subjects experienced profound somnolence, with sleep onset occurring within 30 minutes and lasting approximately 24 hours on average. Extrapyramidal symptoms (dystonia, akathisia) appeared either immediately after administration or with considerable delay; their incidence ranged from 33 to 75%. As with fluphenazine, subjects experiencing deep sedation also reported anxiety, tension, episodes of anger, confusion, and disorientation.[29]
By the mid-1960s it had become apparent that haloperidol no longer met the evolving requirements for a non-lethal chemical agent—either in terms of potency or pharmacological profile. More potent spiropyran-class neuroleptics, such as spiroperidol and 3-methylspiroperidol, were considered as possible successors.
Spiroperidol surpasses haloperidol in antipsychotic potency by several fold; in humans, motor disturbances appear at doses as low as 0.3–0.5 mg.[10]
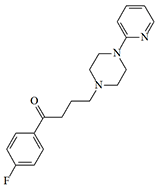 |
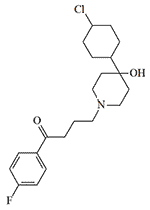 |
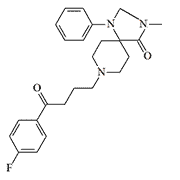 |
 |
| Azaperone | CAR 302,034 (Haloperidol) | CAR 302,089 | EA 5360 |
CAR 302,089 (3-methylspiroperidol) was evaluated as an incapacitant at the Edgewood Research, Development and Engineering Center (ERDEC) in 1965.[17] Relatively little is known about its pharmacological profile: at 0.25 mg/kg it induced light sleep in human subjects; at 20 mg/kg it produced sedation and reduced anxiety and motivation.[9] The potential use of CAR 302,089 in combination with a hallucinogenic benzomorphane was also explored.[2]
Synthesis of spiroperidol derivatives continued at ERDEC from 1966 to 1970, but bioassay results in mice showed no appreciable potency.[24] By contrast, compounds of the EA 5360 type, prepared at FMC Corporation, proved exceptionally active as neuroleptic agents and produced "more pronounced sedation in animals, particularly in primates,"[23] than the spiropyrans synthesized by P. A. Janssen—including spiroperidol and CAR 302,089.[25]
Neuroleptics of Other Chemical Classes
CAR 218,437. Synthesized by Sterling Drug Inc. in 1959.[12] Compounds of this class share a mechanism of action with the neuroleptics; some are more potent than chlorpromazine, but their high toxicity has precluded medical application.[14,19] CAR 218,437 was investigated as a potential incapacitating agent at Edgewood Arsenal in the early 1960s.[15]
 |
 |
 |
| CAR 218,437 | Lu 15-052 | Clozapine |
Lu 15-052 is a highly potent thioxanthene neuroleptic cited as a potential incapacitating agent in Major General N. Antonov's monograph Chemical Weapons at the Turn of Two Centuries (1994). In terms of potency it surpasses phenothiazine-class incapacitants (×4 relative to fluphenazine) and butyrophenone-class agents (×8 relative to haloperidol).[7]
Atypical neuroleptics. For many years it was believed that a neuroleptic free of extrapyramidal side effects was an unachievable goal. That assumption was overturned in 1968 with the synthesis of clozapine, the first atypical neuroleptic—a compound with pronounced sedative-hypnotic activity but virtually no extrapyramidal liability.
No published data appear to exist on the investigation of this pharmacological class as potential incapacitating agents; however, the "knockout" properties of the atypical neuroleptic clozapine are widely exploited by criminal actors to render victims unconscious. Clozapine has now entirely displaced clonidine—previously the agent of choice in Russia and Ukraine—and currently accounts for 99.7% of all reported criminal poisonings. Its toxicological profile is characterized by marked suppression of consciousness with minimal impact on respiration, cardiac function, or blood pressure. Mortality in acute clozapine poisoning is relatively low—0.11%—and occurs predominantly in patients with severe comorbid conditions.[32]
Safety and Drug Interactions
Neuroleptics have one of the widest therapeutic indices of any class of pharmaceuticals. In psychiatric practice, daily doses of fluphenazine may reach 1,200–1,800 mg,[34] a level approximately 25 times higher than the estimated median incapacitating dose (ID50) of roughly 70 mg per person.[11] A case has been reported in which haloperidol was administered at a daily dose of 1,200 mg to a patient with severe cardiac pathology, whereas only 5–10 mg suffices to control acute psychomotor agitation.[38]
Despite an attractive pharmacological profile, neuroleptics have significant limitations that preclude their use as standalone incapacitating agents. The degree of CNS depression they produce is insufficient to achieve complete incapacitation, and extrapyramidal disturbances are inconsistent, delayed in onset, and similarly fall short of rendering a subject fully combat-ineffective. Considerably greater potential for non-lethal applications lies in combinations of neuroleptics with sedative or hypnotic agents—regimens already well established in clinical and veterinary practice.
In anesthesiology, the most widely used combination pairs the butyrophenone neuroleptic droperidol with fentanyl. However, droperidol's duration of action is excessive for an incapacitant,[37] and fentanyl carries a risk of respiratory depression. More promising for non-lethal applications are combinations of neuroleptics (droperidol, haloperidol, olanzapine) with benzodiazepines (midazolam, lorazepam),[35] an approach that reduces the risk of extrapyramidal disorders several-fold.[40]
Experiments conducted in 1973 at the Edgewood Arsenal Biomedical Laboratory demonstrated that phenothiazine neuroleptics substantially potentiate the suppression of mental and physical performance produced by cholinergic deliriants and prolong their duration of action.[39] Additionally, anticholinergic agents such as diphenhydramine and benzatropine prevent or reverse extrapyramidal disturbances.
In 1995, the U.S. Army Chemical Research, Development and Engineering Center (CRDEC) conducted a small study examining the combined effects of the hypnotic etomidate and the neuroleptic azaperone in mice. Azaperone alone did not produce complete immobilization, but when used in combination it permitted a 50% reduction in the etomidate dose required for that effect.[16] In veterinary medicine, azaperone is routinely combined with opioid analgesics for animal immobilization. The combination with etorphine is considered particularly effective and safe: azaperone stimulates the respiratory center and reduces the risk of pulmonary edema—the most serious complication of etorphine-based immobilization.
Use of Neuroleptics in Emergency Psychiadeportationtry and by Law Enforcement
In 1966, studies were conducted in Belgium and the Netherlands on the use of the butyrophenone neuroleptic benperidol in the treatment of antisocial sexual behavior. According to the investigators, the drug "removes the desire to indulge in antisocial forms of sexual behaviour but the ability to function appropriately in the normal sexual environment may be retained."[33]
In the United States, haloperidol has been administered by the Division of Immigration Health Services (DIHS) in cases of resistance to forced deportation, as part of a so-called "pre-flight cocktail." In addition to haloperidol, the formulation includes benzatropine—to reduce the risk of extrapyramidal complications—and lorazepam, which potentiates the sedative effect of the mixture.[27]
Neuroleptics are now routinely employed in emergency psychiatry for the management of psychomotor agitation in schizophrenia, manic psychosis, alcohol delirium, and stimulant, dissociative, or psychotomimetic overdose. Emergency department and psychiatric hospital staff face daily risk from violent and aggressive patients. Neuroleptic use in these settings can serve as a useful approximation for evaluating their suitability as incapacitating agents in scenarios dominated by aggressive behavior—prison riots, ethnic violence, attacks by fan groups or extremist factions, refugee camp disturbances, and similar incidents. Currently, most emergency physicians regard benzodiazepines as the agents of choice,[41] but recent evidence indicates that a neuroleptic–benzodiazepine combination is more effective, faster-acting, and safer than either agent alone.[42]
Prospects for Neuroleptics as Incapacitating Agents
In the view of J. Ketchum, who directed the volunteer experiments at Edgewood Arsenal from 1960 to 1969, neuroleptics are poorly suited as effective incapacitants: they fail to produce complete loss of combat effectiveness, and the emotional indifference induced by these drugs can be overridden in a combat situation by the instinct for self-preservation.[11]
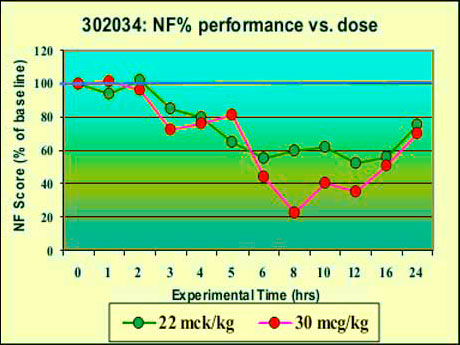
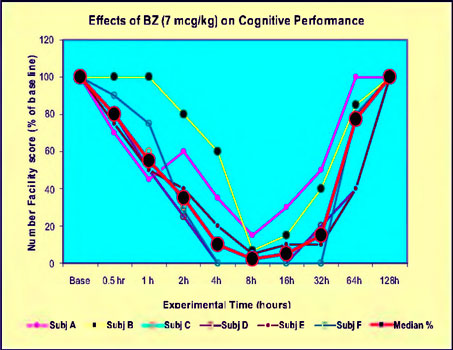
As the graphs illustrate, haloperidol (left) at a dose of 0.030 mg/kg produced a smaller decrement in performance than BZ (right) at 0.007 mg/kg (J. S. Ketchum, 2006).
This assessment, however, is difficult to reconcile with documents attesting to ERDEC's sustained interest in this class of incapacitants, which persisted at least through the mid-1990s. Later volunteer trials involving dozens of subjects were conducted either without Ketchum's participation or after his retirement from service. Russian sources similarly assert that neuroleptics "cause personnel to refuse to carry out orders regardless of the strength of their initial motivation."[8]