Analgesics of Different Groups

Famed drug hunter Paul A. Janssen (1926–2003) discovered analgesics from the groups of Fentanyls, Diphenylpropylamines, Benzimidazolinones and Spirodecanones
Derivatives of Azabicyclononane
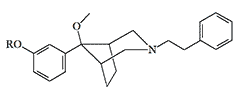
One of the first analgesics in this series, Anazocine, was synthesized in 1969 by Japanese chemists from the pharmaceutical company Sankyo Co. However, it did not possess any exceptional analgesic properties and did not gain widespread use in medicine.[11] But, replacing the N-methyl group of Anazocine with a phenylethyl group and adding a 3-hydroxyl to the benzene ring increases the analgesic activity by hundreds or even thousands of times.[24].
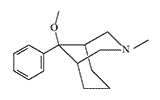
Anazocine |
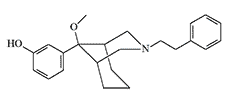
P-7521 |
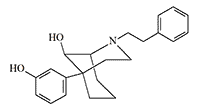
The most active in |
Interest in this group of analgesics gradually decreased until 1982 when scientists in China began actively studying ultrapotent analgesics and rediscovered this drug.
The most studied is azabicyclononane derivative with code P-7521. Depending on the testing method, its activity in rodents and rabbits ranges from X233 to X3860 potency of morphine (see Table 1 under spoiler).[4,12,24,31] In rabbits, compound P-7521 exhibited pain-relieving properties when administered at a dose of 0.00037 mg/kg, with no appreciable respiratory depression even at doses 1200 times higher.[12] It is likely that the reason for this is because P-7521 functions as a μ-receptor agonist with κ-receptor antagonist properties.[3] Like many other opioids, P-7521 has a very high therapeutic index (LD50/ED50) of over 65000 in mice,[24] which does not at all mean that it will be as safe in humans.
| Species of animals | Route | Type of test | Analgesic dose (AD50)* mg/kg |
Analgesic potency (Morphine=1) |
Ref. |
|---|---|---|---|---|---|
| Mice | intraperitoneally | Hot plate test | 0.007 | 1120 | [31] |
| Mice | intraperitoneally | Hot plate test | 0.0066 | 1802 | [4] |
| Mice | subcutaneously | Writhing test | 0.0043 | 233 | [4] |
| Mice | subcutaneously | Haffner’s tail pinch method | 0.003 | 1670 | [24] |
| Rats | subcutaneously | Tail-flick | 0.0017 | 3860 | [4] |
| Rabbits | intravenously | Pain threshold (ear) | 0.00037 | 427 | [12] |
From the peculiarities of pharmacodynamics of azabicyclononanes note a relatively slow achievement of peak effect — 20–60 min after intravenous injection and a too long time of action — about 7 hours.[4] It is worth noting that among azabicyclononanes there are analgesics with a rapid action of 3–8 minutes, but they are less active.[4]
Since 1988, the Edgewood Research, Development and Engineering Center (ERDEC) has been studying incapacitants from the group of azabicyclononanes, and work in this direction continued until at least 1997.[13,14]
Azabicyclononanes. Only preliminary data is available, but results so far indicate real promise for a safe, potent chemical immobilizer for use where an onset of several minutes is acceptable. Problems with CW treaty and controlled substance issues should be much less than with the fentanyls.
All of these substances are solids that have to be disseminated as aerosols for inhalation effectiveness. Thus, a protective mask should serve as an effective countermeasure. Effective antidotes are available for the fentanyls. These antidotes may also be effective against the azabicyclononanes, but we do not have specific knowledge of this.
C. Parker Ferguson, Technical Director ERDEC[2]
In the United States, azabicyclononanes and ultra-short-acting analgesics such as remifentanil were considered the most promising direction in the development of opioid incapacitants.[2]
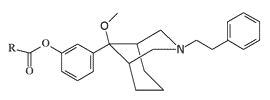
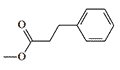
Information about this group of analgesicswas received by the non-governmental organization "Sunshine Project", but the U.S. Navy tried in every possible way to prevent the publication of even the mention of the existence of this group of incapacitants.[29] Therefore, the data on specific substances experimented with in those years remain classified, but the fact that among these azabicyclononane analgesics there are substances with striking activity is beyond doubt. For example, the propionic ester P-7521 is more than 2000 times more active than morphine (see Table 2 under spoiler).[31]
In addition to azabicyclononanes, in China and ERDEC has also studied analgesics from the group of azabicyclooctanes.[14] Also, 7-azanorbornanone congeners of Epibatidine were synthesized and studied at ERDEC.[1]
 |
|||
| Code | R | AD50* | Analgesic potency (Morphine=1) |
|---|---|---|---|
| P-7548 | Ethyl | 0.004 | 2010 |
| P-7617 | Allyl | 0.007 | 1120 |
| P-7528 | cyclo-Pr | 0.009 | 910 |
| P-7556 | Ph | 0.009 | 880 |
| P-7616 | 3-Furyl | 0.010 | 790 |
| — | Benzyl | 0.013 | 620 |
| — | Vinyl | 0.015 | 520 |
| P-7618 | CH2CH2-cyclo-Pentyl | 0.025 | 310 |
| — | i-Pr | 0.04 | 200 |
| Morphine | 7.85 | 1 | |
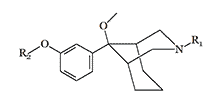 |
|||
| R1 | R2 | AD50* | Analgesic potency (Morphine=1) |
|---|---|---|---|
 |
H | 0.007 | 1120 |
 |
H | 0.008 | 990 |
 |
OCOC2H5 | 0.014 | 570 |
 |
H | 0.022 | 360 |
 |
H | 0.025 | 310 |
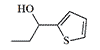 |
H | 0.032 | 250 |
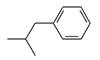
| n-C5H11 | H | 0.054 | 150 |
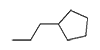 |
H | 0.067 | 120 |
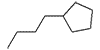 |
OCH3 | 0.155 | 50 |
| Morphine | 7.85 | 1 | |
 |
||
| R | AD50* | Analgesic potency (Morphine=1) |
|---|---|---|
| H | 0.012 | 630 |
 |
0.012 | 630 |
| Morphine | 7.85 | 1 |
* AD50 — Dose in mg/kg causing analgesia in 50% of mice in the hot plate test, intraperitoneally[31]
One of the derivatives of morphane, which is an isomer of 2-azabicyclononane, completely suppressed morphine abstinence at doses of 0.005 and 0.03 mg/kg and was more than 1000 times more potent than morphine.[20] Interestingly, although these compounds were synthesized much later, in the mid-2000s,their research involved experts who developed incapacitants for the US Defense Department in the 60s and 70s.[21]
Knaus compounds
In 1982, Canadian chemists under the leadership of Edward E. Knaus synthesized and studied compounds from the group of piperidylidene-2-sulfonamides. The obtained substances had a structure atypical of opioids but exhibited a pronounced analgesic effect. Compound W-18 in experiments on mice demonstrated fantastic activity — in a dose of only 0.0000037 mg/kg, it caused the same analgesic effect as 0.038 mg/kg of morphine.[5] As a simple calculation shows, the W-18 substance must be at least 10,000 times stronger than morphine.
In N. S. Antonov's monography "Chemical Weapons at the Turn of Two Centuries" (1994), this Compound W-18 is mentioned as a potential incapacitant under the name "Canadian analgesic."[16]
 |
 |
| Compound W-18 | Compound W-15 |
Unfortunately, there have been no additional studies confirming or refuting the high analgesic activity of E. Knaus compounds. In 2012, a substance called W-15, which is different from W-18 only in the absence of a nitro group and was five times more potent than morphine in animal experiments, appeared on sale in e-shops, selling designer drugs. According to reports on recreational forums, the substance had no noticeable effect on humans.
In 2016, after several tragic deaths in Canada caused by W-18 overdoses, the pharmacological properties of the drug were finally seriously investigated.[30] As expected, both drugs W-15 and W-18 are completely devoid of opioid activity.[19]
Bromadol and Thiobromadol
Bromadol (BDPC). In 1979, D. Lednicer and F. Von Voigtlander published a synthesis of a new extremely potent opioid that exhibited analgesic effects in rodents at dose of 0.1 µg/kg.[6]
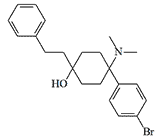 Bromadol |
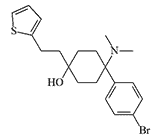 Thiobromadol |
More recent animal studies have shown less analgesic activity — x500 of morphine.[8] This is also confirmed by posts on recreational substance forums, which mention the opioid activity of bromadol for humans already at a dose of 0.025 mg[15] and even 0.01 mg[32] in people with a high tolerance to opioids.
Thiobromadol (C-8813). Substitution of the phenyl group for the thienyl group does not affect physiological activity — thiobromadol is not inferior to bromadol in efficacy[8]. Substitution of p-Br- for m-OH- slightly reduces activity, up to x260-300 compared to morphine.[10]
In April 2013, Canadian police identified bromadol in a batch of confiscated synthetic drugs.[9]
Diphenylpropylamines
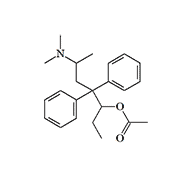
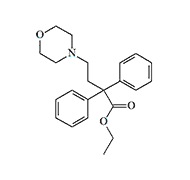
In the 40s and 50s, there were no analgesics much more potent than morphine that could be used in non-lethal chemical warfare. However, some of the synthetic analgesics synthesized in those years were considered by the CIA as potential "knockout" and "anti-interrogation" agents. One of the surviving MKULTRA program documents mentions methadol derivatives: «Some of those compounds produce an extended (36 to 40 hrs) loss of consciousness and reversible paralysis».[17]
 |
 |
 |
| Levacetylmethadol (LAAM) | Dioxaphetyl butyrate X0.1 morphine |
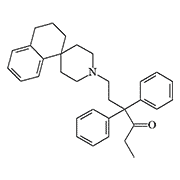
Spirodone X212 methadone |
It seems that this substance is Levacetylmethadol (LAAM), which is known to have analgesic effects for several days after injection. The use of such analgesics to counter harsh interrogation techniques, including torture, has also been considered.[18]
Dioxaphetyl butyrate — is another analgesic of this group, which was of interest to specialists working for the CIA. This drug in humans "produces a rather acute toxic psychosis of two to three days."[18]
Spirodone (R-4066) is a highly potent analgesic belonging to the diphenylpropylamine derivatives. In animal tests, it is 212 times more potent than methadone[39] (in parenteral administration, analgesic doses of methadone and morphine are approximately equal). There is no information on the effect of spirodone on humans.
Derivatives of Benzimidazolinone
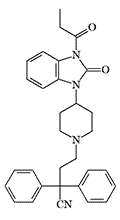
In 1963, P. Janssen synthesized a new opioid analgesic with a benzimidazolinone structure, which was named Bezitramide.[33] The new drug did not have any advantages over other analgesics, took too long to act, and often caused nausea and vomiting. After the death of a 5-year-old child who accidentally swallowed one tablet of Bezitramide, it was withdrawn from the market.[34]
 |
 |
 |
 |
 |
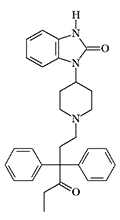
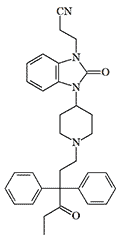
| Bezitramide (R 4845) | R 4837 X1500 pethidine[38] |
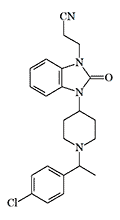
Cyetodezitramide =carfentanil (?) |
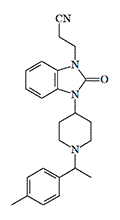
Cychlorphine X270 morphine[35] X2300 pethidine[38] |
X540 morphine[35] |
In 1968, P. Janssen published an article containing formulas and structure-activity relationship data of the most active benzimidazolinones he had synthesized, such as R 4837, and substances known today under the non-official names Cyetodezitramide and Cychlorphine. The morphine-like potency of these benzimidazolinones in mice and rats was higher than that of fentanyl.[38]
In 1984, a group of Chinese scientists from the Shanghai Institute of Materia Medica, who was also working on the synthesis of ultra-powerful analgesics such as 3-methylfentanyl, ohmefentanyl, and azabicyclononanes, became interested in this article. By replacing chlorine with methyl in Cychlorphine they managed to produce an analog with a potency more than 500 times that of morphine.[35]
The addition of a 2-cyanoethyl group to R 4837 further increases its analgesic and euphoric effects. According to a person who has experienced the effects of these opioids, R 4837 is almost equal in activity to 3-methylfentanyl, while Cyetodezitramide is similar to carfentanil in potency.[44]
It is very likely that the purpose of work on benzimidazolinones both in the United States and China was to develop chemical incapacitants. In the United States, the toxicological properties of R 4837, cyetodezitramide, cychlorphine, and other benzimidazolinones were studied at Edgewood Arsenal.[36] Information about these substances or samples of them may have been obtained through Project MKCHICKWIT (1967–1973), a joint Chemical Corps and CIA program to search for potential chemical agents among substances synthesized in laboratories in Europe and Asia.
Like other potent opioids, benzimidazolinones can easily cause a fatal overdose requiring immediate antidote administration. In addition, all substances are too long-acting to be used as incapacitants.
Cychlorphine c late 2023 is offered by online stores selling designer drugs as an opioid four times more potent than fentanyl.[40,46]
Derivatives of Spirodecanone
Opioid agonists from the group of 1,3,8-triazaspiro(4.5)decan-4-one derivatives were first synthesized and studied by P. Janssen in the early 60s.[37] Despite their high morphinomimetic potency, there is very little information on their pharmacological and toxicological properties. The substance R 5260 has binding affinities for μ-opioid receptors almost ten times higher than fentanyl, but in rats, potency of R 5260 and R 6890 (Spirochlorphine) is about the same with fentanyl.[41] In animal experiments, spirodecanes show activity on the level of fentanyl but are very long-acting.
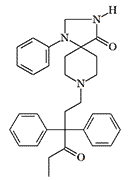 |
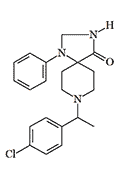 |
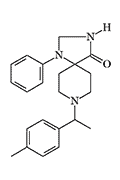 |
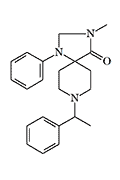 |
| R 5260 | R 6890 X86 morphine[38] |
R 6372 X186 morphine[43] |
X205 morphine[38] |
According to data from specialized websites, R 6890 produces euphoria in humans at a dose two times lower than fentanyl[42]. The activity of R 5260 is on par with 3-methylfentanyl.[42] It is unclear whether these substances can effectively induce anesthesia or immobilization in humans.
Spirodecanones with a butyrophenone chain at the piperidine nitrogen were of much greater interest to both P. Janssen and the US Army. These substances lose opioid activity but have a highly potent antipsychotic effect. Janssen developed the commercial drug Spiroperidol (Spiropitan) for the treatment of schizophrenia, which was briefly marketed in Japan. Incapacitant 3-Methylspiroperidol (CAR 302,089) was tested on volunteers at the Edgewood Arsenal laboratory.
The appearance of information on potent benzimidazolinones and spirodecanones in open sources (1985–1987) indicates a loss of interest in them by American and Chinese developers of non-lethal chemical weapons.[35,47]
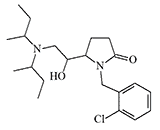 |
Z4349 Another analgesic with an unusual structure for opioids. In animal experiments, it is 320 times stronger than morphine and has a lower potential for physical dependence.[26]. |
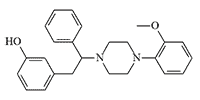 |
Diphenpipenol (MT-45). A derivative of diphenylethylpiperazine, with analgesic activity 100 times that of morphine[27]. Substances in this group are increasingly popular designer drugs and often result in poisoning, with symptoms characteristic of all opioids, such as myosis, cyanosis, apnoea, and deep unconsciousness. In addition, temporary hearing loss, lasting up to two weeks, has been reported in ⅓ cases[28]. |
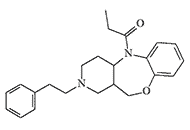 |
Rigid analogs of fentanyl with conformationally restricted 4-anilido group and almost the same antinociceptive properties[23]. |
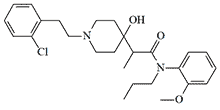 |
Analgesic synthesized by Sandoz in 1982. In animal tests, it showed 30–200 times higher analgesic activity than morphine. Causes mydriasis and, in high doses, CNS depression[22]. |